






Small molecule drug discovery can be hard! Computational approaches can revolutionize the process. But they also have their own unique set of challenges. In this blog post series we're diving into these challenges as well as describing ways you can gain the best of computational methods. If you'd like a live presentation around many of these topics, register for our May 9th webinar on getting started with computational drug discovery!
In our previous post, we introduced docking and virtual screening, powerful tools that help us sift through billions of potential molecules to identify promising lead molecules that can be developed into drug candidates. Despite their successes, these methods have inherent flaws that can lead to expensive failures later in drug development.1, 2 Here we will review where docking and virtual screening fall short – both the bad, and the ugly (Figure 1).
Why Docking and Virtual Screening Haven't Solved Everything Yet
.png)
Challenge #1: Knowing Your Target Protein is Essential
There's a hint of a "chicken and the egg"-style dynamic to protein target location.
To dock a potential drug to a target protein structure, you have to know the structure for the protein first.
Sure, there are a number of traditional methods structural biologists have used to procure protein target structure, including:
- X-ray crystallography
- Cryo-electron microscopy (cryo-EM)
- and nuclear magnetic resonance (NMR)
These are invaluable tools preceding structure-based drug design and virtual screening via docking.
But these methods often fall short for a variety of reasons:
- Limited human time
- Complications in experimental conditions
- Intrinsic properties of proteins that limit the ability to capture all domains or regions
In the end, this means that elements crucial for understanding the biology and disease mechanisms of a given protein are simply missing. Additionally, these methods can fall short resolving structures of disease-relevant mutants.
This lack of complete structural data can limit the effectiveness of computational drug discovery tools.
Machine learning models such as AlphaFold, RoseTTAFold or and ESMfold have helped by generating predicted structures for proteins, but these structures may struggle to model the effect of mutations, predict the correct structure of the binding sites or active sites and often can’t account for biological context like cell membranes.4, 5, 6, 7
Understanding how the target protein works also matters. Drugs are designed to have a specific effect - usually to inhibit or activate a protein’s function. It’s difficult to design a drug to do either of these without understanding what shape a protein takes in inactive or active form (Figure 2). While some of these questions can be answered computationally, old-school wet lab experiments remain crucial to understand the structure-function relationship for each protein.
.png)
Challenge #2: Docking scores and poses are often inaccurate
A major limitation of current docking and virtual screening tools is their inability to accurately predict which molecules will successfully bind to a target site and how they would bind. These are well-known issues of scoring function and sampling. Among the top 1000 molecules predicted to bind by any docking software, only a handful, if any, turn out to be actual binders in experimental validation (Figure 3).8, 9, 10, 11
This is because docking provides little insight on how well molecules actually bind - for decades, there has been little correlation between docking score and actual experimental binding affinity.10, 12, 13, 14, 15, 16, 17 This means that the ranked order provided by a docking or virtual screening of a set of molecules is generally not helpful in prioritizing which molecules to synthesize. These problems exist for even widely-used, highly-cited tools such as Autodock Vina.14, 18
.png)
Beyond measures of whether molecules bind, docking and by extension virtual screening often gets docking poses wrong.21, 22, 23 These docking poses are sometimes used as a starting point for molecular dynamics, or for more rigorous molecular dynamics to more accurately predict binding energy.10, 24, 25 However, if the original pose was incorrectly predicted by docking, FEP is naturally going to further exacerbate this error. In other words garbage in, garbage out: poor binding poses lead to poor molecular dynamics results. This can often mean that prior to molecular dynamics, an expert needs to manually check binding poses of molecules in a binding pocket, limiting the ability to go directly from finding a binder to optimizing it into a lead.
Improving this situation may involve employing an ensemble docking approach in virtual screening, which uses multiple conformations of the protein's binding site. This method can potentially enhance the accuracy of the predictions. However, if the underlying docking and scoring functions are inadequate, this approach could compound the errors, leading to even less reliable results. When docking billion compound libraries, even small errors in scoring function or inaccurate pose prediction docking models, can propagate these errors by prioritizing a ton of false positives among the ranked top thousands of compounds.
This culminates in a significant issue: virtual screening tools often recommend non-binding molecules (false positives) and miss other potentially effective binders (false negatives) (Figure 4). As a result, the industry routinely invests millions of dollars and several months synthesizing thousands of compounds in discovery campaigns, only to find a few viable starting points for further development. And academic teams are routinely surprised that of the 20 molecules they ordered on their limited budget, zero binds the target well (e.g., <10 μM binding affinity).
Challenge #3: Multiparameter Optimization
But a strong binder alone does not make a drug. Ask anyone in drug development what they’re optimizing for most, and they’ll respond “EVERYTHING!” Turning a hit molecule into a drug is a tightrope act of balancing requirements in on-target binding/inhibition capabilities of the compound with off-target binding/inhibition, general toxicity, ADME* , and manufacturability**.24, 26 You want something that will be as effective as possible at intervening in disease, with minimum safety effects and hassle to synthesize (Figure 4).
.png)
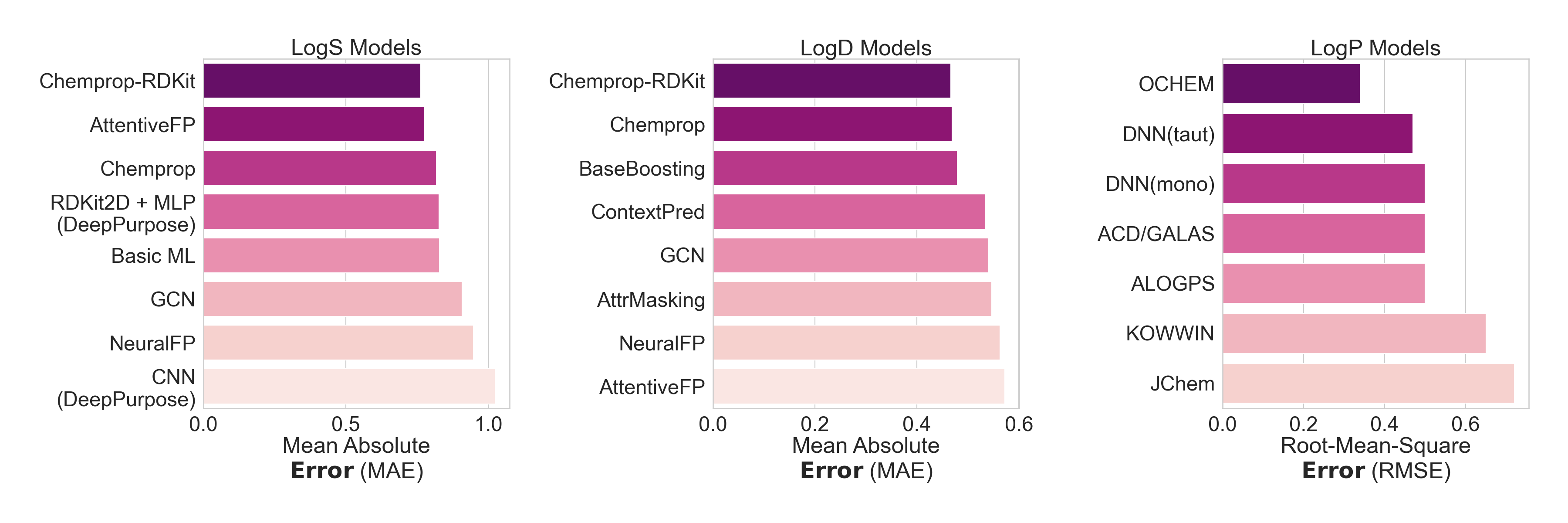
While models have been independently built to predict a wide array of physicochemical properties, few have been integrated cleanly into docking or virtual screening pipelines to enable multiparameter optimization. The models that are available generally have one of two problems: either they are not very predictive (Figure 6), or they are embedded in high-cost software that is not generally accessible. And even with existing tools, a typical journey from hit to optimized lead can take months to years.26, 27
Challenge #4: Maximizing Search Space
With the number of parameters under optimization, docking and virtual screening face a pure numbers problem: your search to find as many needles as possible in the figurative haystack of a molecular library means you’ve got to search a really, really big haystack. Expanding the search space has often shown to increase the chances of discovering promising hits, even for molecules that stray from traditional 'druglike' properties.31, 32
Over the years, chemical synthesis companies like Enamine and eMolecules have constructed ever-larger libraries for screening purposes, growing from millions to billions of compounds. These massive collections explore new chemical spaces with less patent activity and include unenumerated spaces composed of molecular fragments (synthons) and potential reaction pathways. These libraries now number in the tens and hundreds of billions of molecules. However, this expansion brings a significant challenge: conventional docking tools, such as AutoDock Vina, are not designed for high-throughput screening on this scale.33, 34 Managing these colossal libraries demands computational power and time beyond what most organizations can reasonably allocate.
.png)
While virtual screening tools are better equipped to handle these vast libraries, they too face limitations as the size reaches into the billions, often requiring days to weeks to produce results. Moreover, the sheer volume dilutes the probability of identifying effective binders, as the likelihood of discovering an actual binder decreases with the increasing size of the library.
Conclusion
Ultimately, while docking and virtual screening provide a solid starting point, transforming these initial findings into viable drug candidates is a long and iterative process. The inherent limitations of these computational tools mean that they sometimes can lead researchers into costly and time-consuming dead ends. To improve the trajectory of drug discovery, addressing these challenges is crucial. Better predictive tools are needed to streamline the selection of molecules that will eventually go into the clinical trials, aiming to enhance the discovery and development of more effective drugs.
Stay tuned to get a glimpse of what we’ve been building in this space to tackle the critical challenges in docking and virtual screening. You can subscribe to our newsletter here so you don’t miss our beta release.
Footnotes:
* absorption, distribution, metabolism, and excretion
** these are things like solubility, the shelf-life, and the number of synthesis steps to make a molecule
References
- Phillips MA, Stewart MA, Woodling DL, et al. (2018) Has Molecular Docking Ever Brought us a Medicine? Molecular Docking. InTech. Available at: http://dx.doi.org/10.5772/intechopen.72898.
- Walters WP, Wang R. New Trends in Virtual Screening. J Chem Inf Model. 2020 Sep 28;60(9):4109-4111. doi: 10.1021/acs.jcim.0c01009. PMID: 32981325.
- Buttenschoen, M., Morris, G.M., Deane, C.M., 2023. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. http://arxiv.org/abs/2308.05777
- https://www.ebi.ac.uk/training/online/courses/alphafold/validation-and-impact/how-accurate-are-alphafold-structure-predictions/
- AlphaFold and beyond. Nat Methods 20, 163 (2023). doi: 10.1038/s41592-023-01790-6
- Akdel, M., Pires, D.E.V., Pardo, E.P. et al. A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol 29, 1056–1067 (2022). doi: 10.1038/s41594-022-00849-w
- Terwilliger, T.C., Liebschner, D., Croll, T.I., Williams, C.J., McCoy, A.J., Poon, B.K., Afonine, P.V., Oeffner, R.D., Richardson, J.S., Read, R.J., Adams, P.D., 2022. AlphaFold predictions are valuable hypotheses, and accelerate but do not replace experimental structure determination.doi: 10.1101/2022.11.21.517405
- Zhu, T., Cao, S., Su, P.-C., Patel, R., Shah, D., Chokshi, H.B., Szukala, R., Johnson, M.E., Hevener, K.E., 2013. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis: Miniperspective. J. Med. Chem. 56, 6560–6572. https://doi.org/10.1021/jm301916b
- https://www.science.org/content/blog-post/virtual-screening-versus-numbers
- Cutrona, K.J., Newton, A.S., Krimmer, S.G., Tirado-Rives, J., Jorgensen, W.L., 2020. Metadynamics as a Postprocessing Method for Virtual Screening with Application to the Pseudokinase Domain of JAK2. J. Chem. Inf. Model. 60, 4403–4415. https://doi.org/10.1021/acs.jcim.0c00276
- Gimeno, A., Ojeda-Montes, M., Tomás-Hernández, S., Cereto-Massagué, A., Beltrán-Debón, R., Mulero, M., Pujadas, G., Garcia-Vallvé, S., 2019. The Light and Dark Sides of Virtual Screening: What Is There to Know? IJMS 20, 1375. https://doi.org/10.3390/ijms20061375
- Pellicani, F., Dal Ben, D., Perali, A., Pilati, S., 2023. Machine Learning Scoring Functions for Drug Discovery from Experimental and Computer-Generated Protein–Ligand Structures: Towards Per-Target Scoring Functions. Molecules 28, 1661. https://doi.org/10.3390/molecules28041661
- Plewczynski, D., Łaźniewski, M., Augustyniak, R., Ginalski, K., 2011. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J Comput Chem 32, 742–755. https://doi.org/10.1002/jcc.21643
- Wang, Z., Sun, H., Yao, X., Li, D., Xu, L., Li, Y., Tian, S., Hou, T., 2016. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 18, 12964–12975. https://doi.org/10.1039/C6CP01555G
- Pantsar, T., Poso, A., 2018. Binding Affinity via Docking: Fact and Fiction. Molecules 23, 1899. https://doi.org/10.3390/molecules23081899
- https://support.schrodinger.com/s/article/144
- Warren, G.L., Andrews, C.W., Capelli, A.-M., Clarke, B., LaLonde, J., Lambert, M.H., Lindvall, M., Nevins, N., Semus, S.F., Senger, S., Tedesco, G., Wall, I.D., Woolven, J.M., Peishoff, C.E., Head, M.S., 2006. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 49, 5912–5931. https://doi.org/10.1021/jm050362n
- Breznik, M., Ge, Y., Bluck, J.P., Briem, H., Hahn, D.F., Christ, C.D., Mortier, J., Mobley, D.L., Meier, K., 2023. Prioritizing Small Sets of Molecules for Synthesis through in‐silico Tools: A Comparison of Common Ranking Methods. ChemMedChem 18, e202200425. https://doi.org/10.1002/cmdc.202200425
- Bauer, M.R., Ibrahim, T.M., Vogel, S.M., Boeckler, F.M., 2013. Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0 – A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model. 53, 1447–1462. https://doi.org/10.1021/ci400115b
- Jiang, D., Hsieh, C.-Y., Wu, Z., Kang, Y., Wang, J., Wang, E., Liao, B., Shen, C., Xu, L., Wu, J., Cao, D., Hou, T., 2021. InteractionGraphNet: A Novel and Efficient Deep Graph Representation Learning Framework for Accurate Protein–Ligand Interaction Predictions. J. Med. Chem. 64, 18209–18232. https://doi.org/10.1021/acs.jmedchem.1c01830
- Cross, J.B., Thompson, D.C., Rai, B.K., Baber, J.C., Fan, K.Y., Hu, Y., Humblet, C., 2009. Comparison of Several Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 49, 1455–1474. https://doi.org/10.1021/ci900056c
- Agarwal, R., T., R.R., Smith, J.C., 2023. Comparative Assessment of Pose Prediction Accuracy in RNA–Ligand Docking. J. Chem. Inf. Model. 63, 7444–7452. https://doi.org/10.1021/acs.jcim.3c01533
- Shen, C., Hu, X., Gao, J. et al. The impact of cross-docked poses on performance of machine learning classifier for protein–ligand binding pose prediction. J Cheminform 13, 81 (2021). https://doi.org/10.1186/s13321-021-00560-w
- Jorgensen, W.L., 2009. Efficient Drug Lead Discovery and Optimization. Acc. Chem. Res. 42, 724–733. https://doi.org/10.1021/ar800236t
- Salmaso, V., Moro, S., 2018. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 9, 923. https://doi.org/10.3389/fphar.2018.00923
- Hughes, J., Rees, S., Kalindjian, S., Philpott, K., 2011. Principles of early drug discovery. British J Pharmacology 162, 1239–1249. https://doi.org/10.1111/j.1476-5381.2010.01127.x
- Mohs, R.C., Greig, N.H., 2017. Drug discovery and development: Role of basic biological research. A&D Transl Res & Clin Interv 3, 651–657. https://doi.org/10.1016/j.trci.2017.10.005
- TDC.Solubility_AqSolDB
- Ulrich, N., Goss, K.-U., Ebert, A., 2021. Exploring the octanol–water partition coefficient dataset using deep learning techniques and data augmentation. Commun Chem 4, 90. https://doi.org/10.1038/s42004-021-00528-9
- TDC.Lipophilicity_AstraZeneca
- Lyu, J., Irwin, J.J., Shoichet, B.K., 2023. Modeling the expansion of virtual screening libraries. Nat Chem Biol 19, 712–718. https://doi.org/10.1038/s41589-022-01234-w
- Sadybekov, A.V., Katritch, V., 2023. Computational approaches streamlining drug discovery. Nature 616, 673–685. https://doi.org/10.1038/s41586-023-05905-z
- Forli, S., 2015. Charting a Path to Success in Virtual Screening. Molecules 20, 18732–18758. https://doi.org/10.3390/molecules201018732
- Jaghoori, M.M., Bleijlevens, B., Olabarriaga, S.D., 2016. 1001 Ways to run AutoDock Vina for virtual screening. J Comput Aided Mol Des 30, 237–249. https://doi.org/10.1007/s10822-016-9900-9
.png)





.png)

.png)
.png)
.png)

.png)





.png)
.png)

-p-1080%20(1).png)
.png)
.png)
.png)




.png)
.png)


.png)